
Partners Asthma Center Grand Rounds
Jeffrey M. Drazen, M.D.
Pharmacogenetics of Asthma
When one explores the field of pharmacogenetics — or how our genetic make-up influences our responses to drugs — one can consider three different types of effects. First, there are differences in drug metabolism based on genotype, the topic of classic pharmacogenetics. Second, one finds differences in drug targets, our main theme in today’s presentation. Third, genetics can cause differences in drug side effects. An example (not relevant to asthma) of this last type is glucose-6-phosphate dehydrogenase (G6PD) deficiency, the oldest known example of pharmacogenetics. Based on genetic differences in our genes for G6PD, some patients will tolerate drugs such as quinine without any difficulty while others will develop red blood cell lysis as a result of their ingestion.
To derive useful pharmacogenetic information about drug targets, we need two types of information from clinical trial data. We need to know: 1) the variability in the treatment response (or toxicity) among members of a population; and 2) the repeatability of the response within a given member. You would predict that if a response is genetically based, it should 1) occur in some but not all members of a population; and 2) occur in an individual not only once but at many points in time because it is part of his or her fundamental (inherited) make-up.
Variability among members of a population
An example in asthma of finding variance in the treatment response among members of a population comes from a three-way comparison between montelukast, beclomethasone, and placebo. Subjects entered into this trial had mild-to-moderate persistent asthma (FEV1 approximately 65-80% of predicted) and were taking other asthma medicines. After a two-week run-in period when they received placebo, subjects were randomly allocated to receive montelukast, beclomethasone, or placebo for 12 weeks. Overall, the improvement in FEV1 for patients given beclomethasone was on the order of 13-15% and for montelukast on the order of 7-8% (Figure 1). Based on these data, you might reasonably conclude that in this setting beclomethasone is twice as good as montelukast.
Another way to view these same results takes into account population variability. This variability can be viewed by displaying the results as a histogram, with the percentage of patients plotted on the vertical axis and the FEV1 as a percent change from baseline plotted in bins along the horizontal axis (Figure 2). In this example, both patients treated with beclomethasone and patients treated with montelukast demonstrated a large amount of population variability: some patients improved a great deal, some very little, and some deteriorated on therapy. If one considers a 10% or greater increase in FEV1 as a response likely to be associated with appreciable clinical improvement, then approximately 45-50% of patients had clinically meaningful responses. And although the chances of doing better were greater on beclomethasone than montelukast in this trial, they were by no means twice as great.
Repeatability within a given member
Our second question is whether there is repeatability of the response within a given patient. Ideally, what we would really like to know is the hereditability of a treatment response. In a perfect world we would follow families through multiple generations and in each generation test the response of the children at approximately the same age to a particular medication. Even then we would need to ensure that the environments in which the children grew up were relatively stable. Of course, this sort of experiment is unlikely ever to be performed. Instead, we look to the repeatability of the response: in an individual patient, is the improvement (for example, in lung function) seen early on in the experiment sustained throughout the trial? And for another patient, is the deterioration observed at the first follow-up visit maintained at each of the subsequent visits?
Continuing with our theme of the leukotriene pathway, we can examine the repeatability of responses in a data set of patients treated with zileuton, an inhibitor of the enzyme, 5-lipoxygenase. In this randomized, double-blind, placebo-controlled trial of approximately 260 asthmatic subjects, spirometry data were collected after 8 days of therapy (with zileuton or placebo), and again at 36, 64, and 92 days of treatment. If patients had 12% or greater improvement in FEV1 after 8 days of treatment, how likely were they to maintain that improvement on at least 2 of the 3 subsequent visits? Similarly, among non-responders, was the failure to improve observed at 8 days predictive of lack of improvement at subsequent visits? Statistical analysis of these data indicated a reasonable degree of repeatability, on the order of 65-70%.
So, armed with this initial information about leukotriene-modifying drugs — the findings that there is variability in the population response and repeatability in an individual’s response — how can one identify a specific pharmacogenetic effect? There are five key steps to the process: identify a treatment-related biochemical pathway; identify DNA sequence variants in that pathway; associate variable biologic function with a sequence variant; associate the DNA variant with the drug response; and, finally, replicate the observations in an alternative population.
Step 1: Identify a treatment-related metabolic pathway.
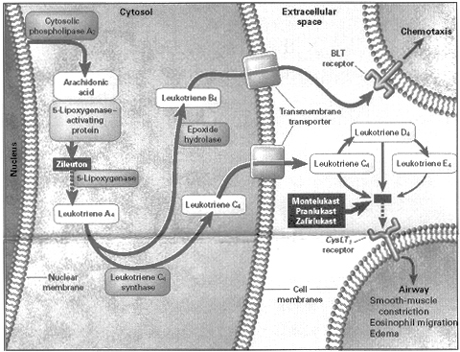
Work done here and at the Karolinska Institute in Sweden beginning in 1969 elaborated the details of the metabolic pathway for leukotriene formation. As shown in Figure 3, the enzyme, cytosolic phospholipase A2, cleaves arachidonic acid from membrane phospholipids. Arachidonic acid is then chaperoned by the 5-lipoxygenase activating protein to the enzyme, 5-lipoxygenase (5-LO). Lipoxygenation of arachidonic acid leads to formation of the unstable intermediate, leukotriene A4 (LTA4). LTA4 becomes the substrate for the enzyme, leukotriene C4 synthase. By addition of glutathione at the C6 position, LTC4 is formed. LTC4 is extruded into the extracellular fluid, where amino acids are excised to form LTD4 and LTE4. LTC4, LTD4, and LTE4 act at the cysteinyl leukotriene (cyst LT1) receptor to induce airway smooth muscle contriction, eosinophil migration, and edema formation.
Step 2: Identify sequence variation in that metabolic pathway
We initially focused our attention on the 5-LO gene, looking for common DNA sequence variants that would lead to changes in the amino acid sequence of the protein, 5-LO. We and others have sequenced the 5-LO gene and found that such sequence variants, although they occur, are too rare (frequency <0.15) to be meaningful. However, just upstream of the translation start site of the 5-LO gene, in a region of the 5-LO gene promoter called the transcription factor binding region, we discovered sequence variants that are more common in the general population. The DNA motif GGG/CCG binds the transcription factor Sp1. In the normal, “wild-type” gene, this Sp1 binding motif occurs 5 times in a row, whereas variants that we found had 3, 4, 6, and 7 repeats in a row of this motif. With this discovery we now had a DNA sequence variant in the enzymatic pathway of interest. Was this a functional variant?
Step 3: Associate biologic variability with the DNA sequence variants
First, we created an oligonucleotide corresponding to our wild-type DNA sequence of interest. In collaboration with Eric Silverman in Tucker Collins’ laboratory here at the Brigham, we were able to demonstrate that this oligonucleotide was capable of binding the transcription factors Sp1 and Egr-1 but not a variety of other transcription factors selected as controls.
We then collaborated with Bengt Samuelsson at the Karolinska Institute to show that this binding event actually drives transcription of the 5-LO gene. Using insect cells that don’t have the capacity to produce Sp1 and Egr-1, and using Sp1 and Egr-1 plasmids to co-transfect these cells, we could demonstrate that binding of these transcription factors resulted in an approximately 35-fold increase in gene transcription. When we eliminated our gene sequence of interest, there was far less induction of transcription. These findings told us that Sp1 and Egr-1 not only could bind to the DNA sequence where we had found genetic variants, but they were also capable of driving gene transcription
The third stage was to determine whether the mutant forms of this DNA sequence transcribed at a different rate than the wild type. When stimulated with Sp1 and Egr-1, the variants with 3, 4, or 6 repeats of the binding motif transcribed at a rate 30-40% lower than the wild type (5 repeats).
Recall that when a patient comes to your office with asthma, you can’t tell whether their asthma might be due to an excess of leukotrienes or substance P or endothelin or neurogenic tone or perhaps a bronchoconstrictor substance that we have yet to recognize. We can’t tell these patients apart; they look phenotypically the same. It’s as if we had only gotten to the stage of diagnosing “anemia” or “pneumonia” without being able to specify the type or specific cause.
One potential way to distinguish types of asthma would be to treat our patients with specific treatments and determine which patients get better. Anti-leukotrienes are very specific treatments, and their specificity can be used to dissect the type of asthma with which we are dealing. If our patients have leukotriene-driven asthma, anti-leukotrienes should make them better. If their asthma is not leukotriene-driven, the anti-leukotrienes should be of little benefit. It follows, then, that if you have a mutant form of the transcription factor binding region of the 5-LO gene, and make less 5-LO enzyme, you might be an anti-leukotriene drug non-responder.
Step 4: Associate the DNA sequence with a drug response
We performed this next step in studying the pharmacogenetics of the leukotriene-modifying drugs at a time when Abbott, the makers of zileuton, were looking for an alternative 5-lipoxygenase inhibitor, one that could be taken once daily and that would be free of hepatic side effects. We were able to obtain samples of DNA from all the subjects enrolled in a 12-week, placebo-controlled, randomized trial of this novel 5-LO inhibitor, ABT761, and stratified the results according to subject genotype. We found that the wild-type DNA sequence (5 repeats) occurred in 81% of the genetic samples that we studied. The deletion alleles (3 and 4 repeats) made up 18.5%, whereas the addition allele (6 repeats) made up less than 1%. Our hypothesis was that subjects with the mutant (deletion) genotypes would transcribe 5-LO protein less effectively and would consequently have a diminished response to the 5-LO inhibitor medication.
Although the study was stopped prematurely when the experimental 5-LO inhibitor proved to have the same event rate for abnormal liver function tests as would have been predicted for zileuton, the results in those subjects who completed the study supported our predictions. Subjects with the wild-type genotype given active treatment with ABT761 had on average an 18% increase in their FEV1 (compared to a 5% increase in those who received placebo). On the other hand, subjects homozygous for the mutant genotypes given the experimental 5-LO inhibitor had no improvement at all (on average, their FEV1 fell by 2-3%). These differences in responses were predicted by subject genotypes and were highly unlikely to have occurred by chance alone. The only shortcoming of these observations is that homozygosity for these mutant deletion alleles is rare, occurring in less than 5% of the population. Thus, these findings can only explain a small portion of the non-responders to anti-leukotriene medications.
Step 5: Replicate the obervations in an alternative population
If we are correct in our hypothesis – that mutant forms of the 5-LO gene cause persons with asthma to be non-responders to the anti-leukotriene drugs because they make smaller amounts of leukotrienes – one would predict that inhibiting the action of leukotrienes with drugs that act at their receptor would duplicate these results. We should be able to replicate the results obtained with the 5-LO inhibitor using leukotriene receptor antagonists, such as montelukast (Singulair) or zafirlukast (Accolate). With cooperation from GlaxoSmithKline, we were able to genotype subjects with mild-to-moderate persistent asthma who enrolled in a comparative trial of fluticasone vs zafirlukast.
Subjects homozygous for the wild-type allele (5 repeats) had significant improvement when treated with zafirlukast (14-15% increase in FEV1), whereas subjects who had no allele for the wild-type gene had a slight decline in lung function when treated with the leukotriene receptor antagonist. In short, in another population of asthmatic patients, we were able to replicate our previous findings of genotype-stratified responses to anti-leukotriene drugs. The overall response to fluticasone was approximately 18-20%. Not addressed in this study are the potential long-term side-effects of the inhaled corticosteroids.
Exploring genetic variants in other proteins within the leukotriene pathway
To explore further the pharmacogenetics of the leukotriene-modifying drugs, we might turn to other enzymes and receptor proteins in the leukotriene pathway. What about genetic variants in LTC4 synthase or in cyst LT1 or cyst LT2 receptors? Genetic studies of LTC4 synthase have found mutations that increase gene transcription. One particular variant, the adenine to cytosine (A-C) transversion at position -444, is relatively common, with a frequency of 0.21. If in fact LTC4 synthase represents the rate-limiting step in leukotriene production – and there is evidence to suggest that it may – then a genetic variant that enhances gene transcription (and hence LTC4 synthase production) would lead to more leukotriene production.
Results to date looking for enhanced LTC4 gene transcription based on these genetic variants have been somewhat inconsistent. Nonetheless, at least two separate groups of investigators have found that when asthmatic subjects homozygous for the a/a genotype are treated with a leukotriene receptor antagonist, they are less likely to improve than are subjects with enhanced LTC4 transcription because of presence of the a/c or c/c genotype. These findings are consistent with the idea that asthmatic subjects who make more leukotrienes are more likely to have a favorable treatment response to leukotriene-modifying drugs.
In addition, we have studied the cyst LT1 receptor gene and found only two sequence variants that cause changes in amino acid sequences of the receptor protein. Both are very uncommon and have no pharmacogenetic consequences or relationship to asthma. More recently, we have examined the cyst LT2 receptor gene. Because of dramatic technologic advances, work that took 1 ½ years for the 5-LO gene and 4 months for the cyst LT1 gene now takes little more than 2 weeks to complete. Of 4 sequence variants in this receptor, one is functional. It is rare in the general population, and we don’t know yet whether there are any biologic consequences.
With these examples I have tried to illustrate the process of pharmacogenetics research. We and other researchers are making our way, one by one, through each step in the leukotriene metabolic pathway, looking for naturally-occuring DNA sequence variants that may make a difference in biologic function. It is likely that the next step will involve examination of the proteins that regulate the expression of these principal proteins, or perhaps the proteins involved in signaling downstream. Our ultimate goal is to use genetic techniques to predict who among our asthmatic patients will respond favorably to leukotriene-modifying drugs and who will not, eliminating the need for a therapeutic trial.
References:
Malmstrom K, Rodriguez-Gomez G, Guerra J, et al. Oral montelukast, inhaled beclomethasone, and placebo for chronic asthma: a randomized, controlled trial. Ann Intern Med 1999; 130:487-95.
Israel E, Cohn J, Dube L, Drazen JM. Effect of treatment with zileuton, a 5-lipoxygenase inhibitor, in patients with asthma. A randomized controlled trial. Zileuton Clinical Trial Group. JAMA 1996; 275:931-6.
Drazen JM, Israel E, O’Byrne PM. Treatment of asthma with drugs modifying the leukotriene pathway. NEJM 1999; 340:197-206.
Figures:
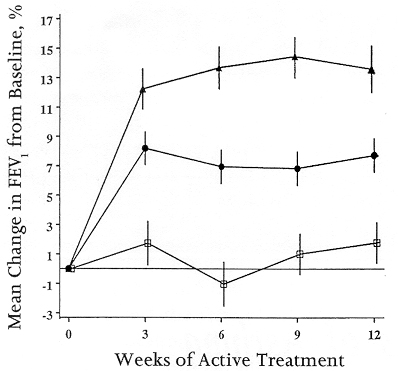
Figure 1: Mean percentage change from baseline in FEV1 during treatment with montelukast 10 mg/day (circles), beclomethasone 200 mcg twice daily (triangles), or placebo (squares). Adapted from Figure 2, ref. #1.
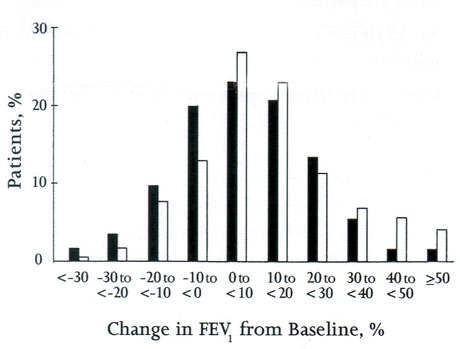
Figure 2: Comparison of distribution of treatment responses (change from baseline in FEV1) for montelukast (striped bars) and beclomethasone (open bars). Adapted from Figure 3, ref. #1.
About the Author:
Dr. Jeffrey M. Drazen is Editor-in-Chief of the New England Journal of Medicine. He is a Professor of Medicine at Harvard Medical School and at the Harvard School of Public Health. He helped to found Partners Asthma Center and remains one of its members.
In the next issue: Asthma in Pregnancy: Controversy and Consensus by Lynda Cristiano, M.D. and Barbara Cockrill, M.D.
Asthma Grand Rounds Bulletin, published between 1997 and 2005, provided brief, written summaries of our early Asthma Grand Rounds presentations.