
Partners Asthma Center Grand Rounds
Elliot Israel, M.D.
Aspirin-Sensitive Asthma
(Originally presented
on
November 10, 2000)
A 42-year-old man presented with asthma and recurrent sinusitis. He had no known allergies, but at age 30 he developed nasal polyps with loss of his sense of smell and taste. At approximately the same time he experienced an asthmatic attack triggered by ingestion of aspirin, and he was told that he had “aspirin allergy.” Several years later, by accident he again took an aspirin-containing product, precipitating severe wheezing and an emergency room visit for treatment of his asthma.
Over the last several years, his asthma has been very active, interfering with his daily activities and his work. At the time that we first met him, his asthma treatment consisted of high-dose inhaled steroids, oral theophylline, an antihistamine and decongestant, nasal steroid, and frequent inhaled beta-agonist (1-2 canisters per month). Three-to-four times each year he needed a burst of oral steroids for control of his symptoms. He had undergone nasal polypectomy 2 years earlier for his recurrent sinus infections and persistent nasal congestion, but his nasal polyps recurred several months thereafter.
With adjustment of his medications, we were able to improve his lung function to achieve an FEV1 of 75% of predicted. He enrolled in a research experiment involving leukotriene inhibitors. As part of the study, he underwent an inpatient aspirin challenge, involving graded increases in the dose of aspirin that he swallowed, with careful monitoring of his pulmonary function as well as urinary leukotriene excretion. In response to a relatively small dose of aspirin, he developed wheezing along with lip and tongue swelling. His FEV1 fell by 36% and his urinary leukotriene E4 level rose from a baseline of 300-500 pg/mg creatinine (normal: 50-100 pg/mg creatinine) to 1500 pg/mg creatinine (normal: no change in response to aspirin).
As part of our experiment, he was treated for one week with the 5-lipoxygenase inhibitor, zileuton (Zyflo). When he returned for his follow-up study, he reported to us that he needed his beta-agonist for relief of symptoms less often, and that for the first time in several years he had recovered his sense of smell.
The first report of a patient with asthma triggered by aspirin appeared in the literature in 1902 (Hirschberg). In 1968 Samter and Beers described what they called “triad asthma”: the syndrome of asthma, aspirin intolerance, and nasal polyposis. Since then “triad asthma” has sometimes been referred to as “Samter’s syndrome.”
The prevalence of aspirin-sensitivity among persons with asthma is difficult to ascertain with certainty. The reported prevalence of 5-30% among adult asthmatics is probably overinflated, given the following scenario. A patient with asthma develops a viral respiratory tract infection with headache and muscle aching. He takes aspirin for relief of his discomfort, and his asthma deteriorates in the setting of a viral bronchitis. It is easy to imagine that this patient might be labeled as having aspirin-sensitive asthma, with a physician thinking that aspirin had caused his worsening, although the true cause of his deterioration was the viral respiratory infection. It is likely that the true prevalence of aspirin-sensitive asthma is 5% or less. Aspirin-sensitive asthma often presents as difficult-to-control asthma, and among severe asthmatics its prevalence is as high as 20%. If you have asthma and nasal polyps, your chance of having aspirin sensitivity increases to 20-30%. The associated rhinosinus disease frequently precedes aspirin intolerance by several years, and it can be the patient’s primary complaint.
Aspirin sensitivity is rare among children (less than age 20). Aspirin-sensitivity is acquired – patients have typically taken aspirin before without incurring asthmatic reactions – with an age of onset between 30 and 50 years. Nasal polyps are likewise uncommon in children with asthma. The combination of wheezing and nasal polyps in a child should make you think of cystic fibrosis, not asthma and aspirin sensitivity.
Aspirin sensitivity, not allergy
Compared to a typical allergic reaction (e.g., to peanuts), the onset of symptoms following aspirin ingestion is relatively slow, on the order of 30 minutes to 2 hours. Asthmatic symptoms are typically preceded by nasal congestion and rhinorrhea. Some patients will also manifest with gastrointestinal distress, including abdominal pain, diarrhea, and rarely bleeding. Occasionally patients experience angioedema in reaction to the aspirin. At one time it was thought that aspirin sensitivity was also associated with a tendency to tartrazine-sensitivity (FDC yellow dye #5) and even to an intolerance of hydrocortisone, but neither of these purported associations is true.
Characteristic of aspirin-sensitive asthma is a cross-reactive intolerance of non-steroidal anti-inflammatory drugs (NSAIDs). Aspirin sensitivity is not a true allergy; it is not an IgE-mediated, immediate hypersensitivity where an antibody recognizes a specific epitope. Although aspirin and the various NSAIDs share the property of inhibiting the enzyme, cyclooxygenase, they are all structurally quite dissimilar. They inhibit cyclooxygenase at a variety of different binding sites, and when examined by three-dimensional modeling, they have distinct chemical structures. What they share in common is the biochemical property of cyclooxygenase inhibition, and their potency in triggering aspirin-sensitive asthma is directly proportional to their in vitro degree of inhibition of the cyclooxygenase isoenzyme 1 (COX-1). Analgesics with relatively weak cyclooxygenase inhibition, such as salsalate (Disalcid) or choline magnesium trisalicylate (Trilisate), are far less likely to cause a reaction in people with aspirin-sensitive asthma. Acetaminophen (Tylenol) is also in this latter category, although at doses of 1000 mg or more, as many as 1/3 of aspirin-sensitive patients will show some reaction to acetaminophen.
Aspirin-sensitive asthma is related to inhibition of cyclooxygenase and, as we shall see, overproduction of leukotrienes. As you know, these two chemical pathways are interrelated (Figure 1). Arachidonic acid, released from cell membranes by the action of phospholipases, can be metabolized by the 5-lipoxygenase enzyme to form the cysteinyl leukotrienes, LTC4, LTD4, and LTE4. These leukotrienes are potent bronchoconstrictors, and they cause recruitment of eosinophils. Other 5-lipoxygenase products of arachidonic acid include LTB4, a neutrophil chemoattractant, and 5-hydroxyeicosatetraenoic acid (5-HETE), a mucus secretagogue. On the other side of the equation, arachidonic acid can be metabolized by cyclooxygenase to form prostaglandins and thromboxanes. So, with elucidation of the metabolic pathway for formation of leukotrienes came speculation that inhibition of cyclooxygenase by aspirin or NSAIDs might shunt arachidonic acid metabolism toward overproduction of leukotrienes, or might lead to decreased production of prostaglandins that have regulatory influence on the 5-lipoxygenase pathway.
Excess leukotriene production
In the early 1990s came evidence from the laboratory of Dr. Tak Lee in London that there is in fact over-production of leukotrienes in patients with aspirin-sensitive asthma when they are given an aspirin challenge. His study compared the responses of patients with aspirin-sensitive and aspirin-tolerant asthma to methacholine and aspirin challenges given on separate days. Outcomes included changes in lung function and production of urinary leukotrienes. At baseline, patients with aspirin-sensitive asthma had, on average, higher levels of urinary leukotrienes (Figure 2). In both groups, in response to bronchoconstriction induced by methacholine, lung function fell but urinary leukotrienes remained unchanged. With aspirin ingestion, patients with aspirin-tolerant asthma had no significant changes in lung function or leukotriene production. However, patients with aspirin sensitivity reacted to oral aspirin challenge with a fall in lung function and a dramatic rise in urinary leukotriene levels.
These findings suggested that if one could block the production or action of leukotrienes, particularly the cysteinyl leukotrienes, it might be possible to abrogate the effect of aspirin and NSAIDs in aspirin-sensitive asthmatics. We conducted such a study using the lipoxygenase inhibitor, zileuton. First we documented the presence of aspirin sensitivity with a double-blind aspirin (or placebo) challenge, using graded doses of aspirin to produce a bronchoconstrictor response. In this way we found that many patients who believe that they are aspirin sensitive are in fact not; and we identified the minimal (threshold) dose of aspirin needed to trigger significant airflow obstruction (>20% fall in FEV1) in our aspirin-sensitive patients.
Patients in this study were treated for one week with the 5-lipoxygenase inhibitor, zileuton, or with placebo. At the end of the week, they were given the threshold dose of aspirin previously identified to cause a significant reaction. We observed them for 6 hours after aspirin ingestion, monitoring lung function and leukotriene production, along with nasal and gastrointestinal symptoms and angioedema.
Pre-treatment with zileuton significantly reduced leukotriene production, as evidenced by a decrease in urinary leukotriene E4 levels. Urinary leukotriene levels were reduced, but not eliminated. Was a reduction of this degree sufficient to impact the clinical response? It was. As shown in the figure (Figure 3), the fall in FEV1 observed after ingestion of aspirin was nearly totally blocked by zileuton pre-treatment. Also, the nasal, GI, and dermal reactions were eliminated in the zileuton-treated group.
This and similar studies have demonstrated the critical role of leukotrienes in causing the airway and other reactions observed in aspirin-sensitive asthma after ingestion of a cyclooxygenase inhibitor. Note, however, that they do not suggest that treatment with leukotriene blockers make it safe for aspirin-sensitive patients to take aspirin or NSAIDs. Leukotriene inhibitors have been shown to block the response to a threshold dose of aspirin, for instance 30 mg. They might well not protect against a larger dose, such as two tablets of aspirin (650 mg). For patients with aspirin-sensitive asthma, the best strategy remains abstinence: careful “bottle reading” and strict avoidance of aspirin, aspirin-containing remedies, and NSAIDs.
Potential pathobiologic mechanisms
What do we know of the cellular and biochemical abnormalities that distinguish aspirin-sensitive from aspirin-tolerant asthmatics? Mast cells and eosinophils are the principal sources of leukotriene production. Using tryptase release as a marker of mast cell activation, it is possible to show that aspirin ingestion causes mast cell activation in aspirin-sensitive asthma. Tryptase levels rise in the nasal washings of aspirin-sensitive asthmatics after aspirin challenge, but not in control subjects. Mast cell activation leads to release of interleukin 5, a potent chemoattractant for eosinophils. Analysis of bronchial biopsies and bronchoalveolar lavage fluid has demonstrated that persons with aspirin-sensitive asthma have increased levels of IL-5 and on average 3-4 times more eosinophils in their airways than persons with aspirin-tolerant asthma.
Clearly, the cellular machinery for leukotriene production is present in persons with aspirin sensitivity, and enzyme abnormalities at any of a number of different steps along the pathway of leukotriene production might result in their excess formation, from 5-lipoxygenase, to 5-lipoxygenase activating protein, to leukotriene C4 synthase, to leukotriene A4 hydrolase. Alternatively, one could postulate that more arachidonic acid substrate is made available or that there is decreased inhibitory influence from one of the prostaglandins.
Evidence to date points to an up-regulation of leukotriene C4 synthase in patients with aspirin sensitivity. Compared to asthmatics without aspirin sensitivity (and to normals), aspirin-sensitive asthmatics have increased levels of leukotriene C4 synthase and a greater percentage of activated eosinophils that stain for this enzyme. Furthermore, the level of leukotriene C4 synthase activity in bronchial biopsies correlates with the degree of aspirin sensitivity (as tested using inhaled lysine aspirin). Furthermore, the level of leukotriene C4 synthase in bronchial biopsies also correlates with the amount of leukotrienes retrieved on bronchoalveolar lavage.
Why there occurs an (acquired) upregulation of leukotriene C4 synthase in aspirin-sensitive asthma remains uncertain. Potential areas that are currently being explored include genetic polymorphisms in the promoter region of the leukotriene C4 synthase gene, genetic associations with HLA phenotypes (HLA-DR11), and deficient production of prostaglandin E2. A role for prostaglandin E2 has been suggested by the finding of decreased levels of PGE2 in nasal polyps from patients with aspirin sensitivity, and the ability to block aspirin-induced bronchoconstriction (and leukotriene production) with administration of inhaled PGE2.
Management of aspirin-sensitive asthma
Management of aspirin-sensitive asthma is based first and foremost on avoidance of aspirin and NSAIDs. For analgesia, narcotics are safe to use. For anti-inflammatory effects, salsalate and choline magnesium trisalicylate can be tried. Preliminary reports suggest that the cyclooxygenase 2 inhibitors such as celecoxib (Celebrex) and rofecoxib (Vioxx) are tolerated in aspirin-sensitive asthma. However, their selectivity is dose dependent, so that at increasing doses COX-1 inhibition begins to be observed and asthmatic reactions can be precipitated.
Leukotriene modifiers are indicated to treat aspirin-sensitive asthma. They have been shown to improve lung function even among patients already receiving corticosteroids for their asthma; and patients on high-doses of inhaled steroids can successfully reduce their dose with addition of a leukotriene modifier. They will also lessen the harmful reaction to inadvertent ingestion of a COX-1 inhibitor.
Aggressive treatment of rhinosinusitis is an important component of therapy. Often nasal and sinus symptoms dominate the clinical presentation in aspirin-sensitive asthma. High-dose nasal steroids (2-4 sprays of a high-potency steroid administered twice daily) may help to shrink nasal polyps; sometimes oral steroids are needed for at least temporary relief. Nasal polypectomy usually provides only temporary benefit. Nasal polyps will typically regrow after surgery, sometimes in a matter of weeks. At times nasal polypectomy acts as a “debulking” procedure, making possible topical administration of nasal steroid sprays.
Occasionally we turn to aspirin desensitization for treatment of aspirin-sensitive asthma. We begin by administering a very small dose of aspirin, anticipating that a mild reaction will be elicited. With repeated administration of aspirin, gradually the dose is escalated until the target (e.g., 325 mg) is achieved. With each dose, some reaction can be expected. In effect, the process leads to depletion of mediators due to recurrent stimulation and activation of the effector cells. The benefits of successful desensitization include tolerance of aspirin, decreased leukotriene production, improved nasal symptoms, and probably improved control of asthma. The disadvantages include the need for a several-day hospitalization to complete the procedure, the risk of inducing a severe reaction, and the need to continue to take aspirin on a daily basis. Omission of aspirin for 2-3 days can lead to loss of tolerance and the need to repeat the desensitization process all over again.
References:
1. Samter M, Beers RF. Intolerance to aspirin: clinical studies and consideration of its pathogenesis. Ann Intern Med 1968; 68:975-983.
2. Christie PE, Tagari P, Ford-Hutchinson AW, et al. Urinary leukotriene E4 concentrations increase after aspirin challenge in aspirin-sensitive asthmatic subjects. Am Rev Respir Dis 1991; 143:1025-9.
3. Israel E, Fischer AR, Rosenberg MA, et al. The pivotal role of 5-lipoxygenase products in the reaction of aspirin-sensitive asthmatics to aspirin. Am Rev Respir Dis 1993;148:1447-1451.
4. Drazen JM, Israel E, O'Byrne PM. Treatment of asthma with drugs modifying the leukotriene pathway. N Engl J Med 1999; 340:197-206.
Figures:
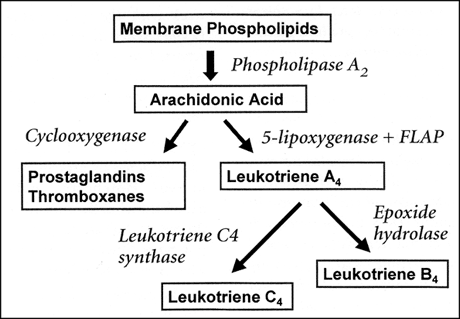
Figure 1: Metabolic pathways of arachidonic acid
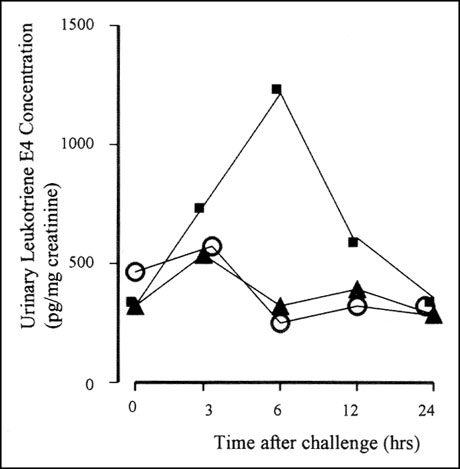
Figure 2: Urinary leukotriene levels in aspirin-sensitive asthmatics after challenge with aspirin (squares), methacholine (triangles), or an oral placebo (circles).
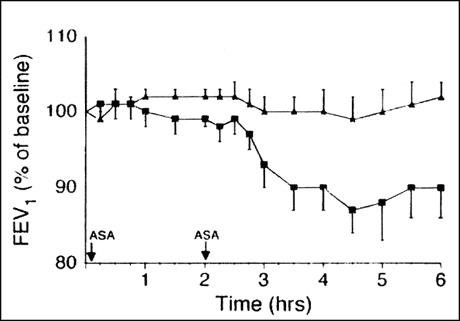
Figure 3: Effect of aspirin challenge on lung function (FEV1) in aspirin-sensitive asthmatics pre-treated with the 5-lipoxygenase inhibitor, zileuton, (triangles) or placebo (squares).
About the Author:
Dr. Elliot Israel is the Director of the Asthma Research Center at Brigham and Women’s Hospital, where he also serves as Medical Director of Respiratory Therapy. He is an Associate Professor of Medicine at Harvard Medical School.
In the Next Issue:
“Nitric Oxide as a Marker of Inflammation in Asthma” by Anthony F. Massaro, M.D.
Asthma Grand Rounds Bulletin, published between 1997 and 2005, provided brief, written summaries of our early Asthma Grand Rounds presentations.