
Partners Asthma Center Grand Rounds
Jeffrey J. Fredberg, M.D.
Basic Mechanisms of Asthma:
Airway Smooth Muscle Function
As discussed by Dr. Ann Woolcock in a recent "Asthma Grand Rounds" presentation (see Asthma Grand Rounds Bulletin, Vol. 3, No. 2, Spring, 2000), asthma can be defined as a condition in which "airways narrow too much and too easily, causing airway closure and symptoms." Airways that narrow too much and too easily have the property that we call airway hyperresponsiveness. Dr. Woolcock presented data from her study of Australian school children that made clear that airway hyperresponsiveness, atopy, and symptoms (recent wheezing) are distinct entities and are not necessarily coincident. You can find people who are atopic but are not airway hyperresponsive and, similarly, people who have airway hyperresponsiveness but are not allergic. It is the intersection of these three properties, about 8% of the general population in Dr. Woolcock's study, that we call asthmatic.
I would like to focus on the subject of airway hyperresponsiveness and to ask the question, what causes airway hyperresponsiveness? To answer that question, I will pose another question: When airway smooth muscle is activated, what sets the final muscle length? Smooth muscle is wrapped helically around the airway, and when activated the muscle shortens, the airway coils inward, the epithelium is thrown into folds, and the airway lumen becomes compromised. In the simplest model of airway smooth muscle function, two key factors set the final muscle length: the isometric force-generating capacity of the muscle on the one hand, and the static load against which that muscle must shorten. The static load is determined by the elasticity of the airway wall, that is, by how stiff the wall is, and by the tethering forces of the surrounding lung parenchyma, which exerts substantial forces to keep the airway open.
It was Dr. Susan Gunst who first suggested that models like these may be incomplete because they fail to take into account certain dynamic properties of the muscle that come into play during tidal respiration. Let me share with you the conclusion of my talk at the beginning. In a system where muscle is activated and given enough time to complete its shortening, the muscle comes to a static equilibrium, with the final muscle length called the static equilibrium length. I am going to show you evidence to suggest that if we take that very same system and do nothing more than impose upon it small force fluctuations ( F) such as are associated with quiet tidal breathing, this system will become dynamically equilibrated with the muscle at a final length that is substantially greater than the static equilibrium length. These force fluctuations exert a bronchodilatory effect and a big one. The notion, then, is that force fluctuations, even if they have a mean value of zero, can substantially bias the final smooth muscle length. We call this phenomenon "fluctuation-driven muscle lengthening." I am going to try to convince you that this phenomenon — or more specifically, its failure — may be the central feature that accounts for airway hyperresponsiveness.
We performed the following experiment using bovine tracheal smooth muscle, although it doesn't matter which species of animal you choose. We set the initial muscle length to the length where the force that it generated was optimal (F0); we stimulated it maximally with a high concentration of acetylcholine; and then we let that muscle shorten against an isotonic load of about 1/3 of F0 (that is, a load in the physiologic range). We waited until the muscle completed its shortening and came to its static equilibrium length. Then, we imposed on that muscle small force fluctuations, F, with a mean value of 0 at frequency in the breathing range (0.2 Hz or 12/min). We measured the final muscle length, meaning the average value of the muscle length over the breathing cycle.
The results (Figure 1) show the final muscle length for force fluctuations ( F) ranging from 4-32% of F0. With very small force fluctuations (4% of F0) nothing happens, but with larger force fluctuations the muscle becomes dynamically equilibrated at a length that is substantially greater than the static equilibrium length. The bigger the force fluctuation amplitude, the longer the length at which the muscle equilibrates. If we then reduce the force fluctuation amplitude, the muscle reshortens.
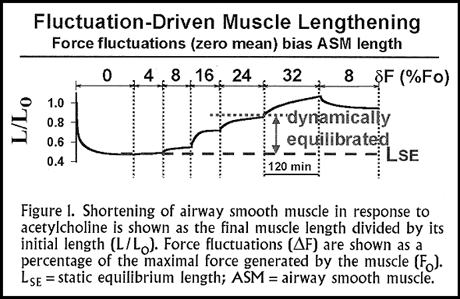
An important point here is that the reshortening of the muscle that occurs as smaller force fluctuations are applied never gets back to the same length as initially seen at that same force fluctuation. This is an important effect reflecting the history of tidal loading. It is important because the stiffness of the muscle reflects the number of actin and myosin interactions. As we increase the magnitude of the force fluctuations, the muscle becomes progressively less and less stiff as a result of fewer and fewer actin-myosin interactions. Also, with increasing force fluctuations there is a higher rate of turnover of these actin-myosin interactions; the rate of actin-myosin crossbridge cycling increases.
With small force fluctuations ( F<8% of F0) applied to the muscle, its final length is virtually the same as its static equilibrium length. The muscle is very stiff or essentially "frozen." With force fluctuations greater than the threshold of approximately 8% of F0, the muscle length gets progressively longer, the phenomenon that we call fluctuation-driven muscle lengthening. A relationship like this one immediately makes one ask, what magnitude of force fluctuations are applied under conditions of normal breathing? The amplitude of force fluctuations imposed on airway muscle by the action of quiet tidal breathing is somewhere between 8 and 16% of F0. With deep inspirations -- and we all take deep inspirations at the rate of about 10 times per hour -- we operate at approximately 24% of F0.
These observations raise an interesting possibility, to which we will return later. Is it possible that normal individuals enjoy plenty of fluctuation-driven muscle lengthening that keeps their airways open when airway muscle is activated, whereas persons with asthma have reduced force fluctuations acting on their airway muscle, allowing them to contract to shorter final lengths, causing bronchoconstriction. For example, we know that in asthma there is thickening of the peribronchial adventitia. This thickened adventitia would reduce the recoil effect that the surrounding lung parenchyma exerts on the airway, thereby decreasing the force fluctuations acting on the muscle.
Actin-Myocin Crossbridge Cycling
What accounts for this phenomenon of fluctuation-driven muscle lengthening? I need to discuss some basic smooth muscle biophysics and introduce the concept of perturbed equilibria of myosin binding. The prevailing concept of cycling-rate regulation for smooth muscle is the latch hypothesis from Hai and Murphy. Myosin is the basic motor protein in smooth muscle. With activation of muscle, myosin becomes phosphorylated by a complex of calcium, calmodulin, and myosin light chain kinase. Only in this phosphorylated state can myosin combine with actin to form actomyosin or so-called rapidly cycling crossbridge. The unique contribution of the latch hypothesis was to suggest that this actin-myosin crossbridge can be the substrate for a phosphatase that can dephosphorylate the myosin while it remains attached to the actin forming a new species, called the slowly cycling latchbridge. It is called that because its rate of detachment in the dephosphorylated state is about 10 times slower that the rate of dissociation when phosphorylated.
Since this concept was first introduced, we know a lot more about it. For example, we now know that there are kinases other than myosin light chain kinase that can phosphorylate myosin (for example, Rho-kinase); and we also know that the dephosphorylation step is a regulated process, also regulated by Rho kinase.
The question that I wanted to ask is, what are the implications of this latch hypothesis for airway mechanics? The answer is made complicated because the latch regulatory scheme just described gets played out in three-dimensional geometry. From the sliding filament model of Huxley, we find myosin in one of four possible states: unphosphorylated unattached (to actin), phosphorylated unattached, phosphorylated attached, or dephosphorylated attached. Myosin can be in any one of these four states and in fact cycles between them. The rate constants that govern cycling between these states are sensitive to the strain of the crossbridge. As the actin filament moves relative to the myosin backbone, as would occur with breathing, it strongly modulates the rates at which myosin cycles between its four possible states. This model provides a mechanism whereby mechanical events (sliding of the actin filament relative to the myosin backbone) are coupled to biochemical events (phosphorylation and dephosphorylation of myosin in attached or unattached states).
The next question to ask is: Are these mechanisms sufficient to account for the observations regarding fluctuation-driven muscle lengthening that I have shown you? We took a quantitative approach using mathematical modeling to compute the implications of Huxley married with Hai and Murphy. We calculated the mechanical, metabolic, and molecular implications, and I will share with you an abbreviated version of our conclusions. As the actin filament moves back and forth just a little bit with tidal strains, it causes the myosin to detach much sooner than it otherwise would have. It reduces the myosin duty cycle. The fraction of time that myosin is attached is reduced to only 20% of what it would be ordinarily, and therefore force goes down and the muscle lengthens.
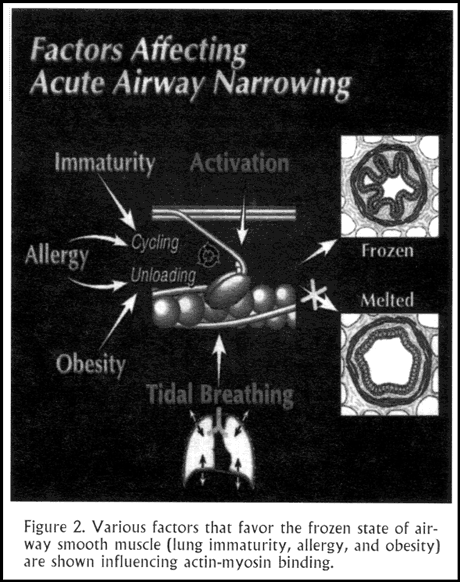
Here is an example: During an isometric contraction, where there are no fluctuations, the contracted muscle comes to a condition with a particular distribution of myosin between attached and detached states, referred to as the static binding equilibrium. In fact, these states are cycling back and forth, attaching and detaching all the time. They are cycling very slowly. It is a cold or "frozen" molecular state: the force is high, but the cycling is very slow. The muscle is stuck in the latch state. From recent physiologic studies we know that in this molecular state, airway muscle in normal animals and humans can generate sufficient force to close every airway in the lung completely (Figure 2).
Perturbed Equilibria of Myosin Binding
In the presence of length fluctuations that would be associated with breathing, there is a perturbed eqilibria of myosin binding, however. Fewer crossbridges are attached, and they are cycling at a very high rate. This is a "hot" molecular.state. Even though we are dealing with the same muscle and the same degree of muscle activation, in the presence of length fluctuations one finds significantly less force generated, fewer bridges attached, and a higher cycling rate. This perturbed equlibria of myosin binding accounts for fluctuation-driven muscle lengthening.
The quantitative analysis from first principles of myosin dynamics, combining the models of Huxley and Hai and Murphy, predicts a threshold effect on final muscle length at fluctuations of approximatley 8% of F0, and above 8% a divergence between fluctuation-driven muscle length and static muscle length, just as we observed. Interestingly, myosin dynamics cannot explain the effect of loading history that we observed. An alternative, plastic phenomenon in muscle must explain this behavior. Clearly there is an advantage to this plasticity in terms of greater muscle length and decreased airways resistance, but the mechanism is not known.
We compared the bronchodilatory effect of force fluctuations and isoproterenol. Muscle was activated with a high concentration of acetylcholine and allowed to come to a plateau of force generated. We then exposed the muscle to a high concentration of isoproterenol or to small fluctuations of muscle length, comparable to those seen with tidal breathing. The response to tidal stretches was much more prompt, and both interventions were roughly equipotent in causing the muscle to relax. This observation indicates that breathing exerts a potent bronchodilator effect. In asthma this mechanism fails, however, although we don't yet know why.
To understand airway narrowing in asthma, we have to consider three widely disparate lengths all at the same time. First, at the organ level we think about fluctuations of transpulmonary pressure. It is these fluctuations in transpulmonary pressure that expand the lungs and have us breathe. Second, these fluctuations in transpulmonary pressure get transmitted to the level of the airway by means of parenchymal tethering of the airway. They cause airway smooth muscle to go through tidal stretches. And third, these force fluctuations acting on the muscle lead to perturbed equilibria of myosin binding. Those perturbed equilibria of myosin binding tend to keep the muscle long and compliant and, by reducing airway resistance, help to facilitate breathing.
These events are tightly coupled across three widely disparate length scales. Taken together, the system as a whole is dynamically equilibrated and it is dissipative, meaning that it requires energy pumped into it by the muscles of the chest wall. In addition, it is a self-reinforcing system. Events at each level tend to facilitate the events at the next lower or higher level. Because of this self-reinforcing nature, stretch of the airway smooth muscle tends to act as its own catalyst.
For airway muscle to remain in a melted or non-frozen state, it is dependent on energy input from the chest wall and the efficient transmission of these forces down to the level of the molecule. These melted states can be described as conditionally stable. The whole system is precariously perched, and it would be easy for it to go awry. For instance, if we took away the tidal stretches from the chest wall or uncoupled the chest wall from the airway muscle due to thickening of the peribronchial adventitia, this dynamically equilibrated system might collapse down the pathway where the muscle would get short and stiff. At the molecular level, the muscle would go into the latch state. It would become so stiff and frozen that it would be refractory to the effects of force fluctuations at the organ level.
If this system is truly perched delicately at the edge, then even a small intervention should precipitate a large change in airway resistance. A relatively modest intervention could cause the system to flip between one state and the other. A simple example would be to test the effect of eliminating the deep inspirations that we normally take approximately 10 times per hour. This experiment was performed by Dr. Peter Pare and his coworkers, based on an earlier protocol (with the same results) done by Dr. Sol Permutt. They took healthy people who were neither atopic nor asthmatic. They measured airway reactivity using incremental doses of inhaled methacholine to a maximal concentration of 16 mg/ml. As one would predict, they found only small (approximately 10%) decreases in the one-second forced expiratory volume (FEV1) at the highest dose of methacholine, consistent with normal airway reactivity.
They then had these same subjects return on a separate day to repeat the methacholine challenge with the only difference being that they were prohibited from taking deep inspirations. They breathed at the same lung lung and with the same size tidal breaths as they had on the previous day, but made no deep inspirations, yawns, or sighs. The researchers found that under these circumstances, airway reactivity changed significantly, with a maximal decrease in FEV1 of approximately 30-35%, a response that one would ordinarily characterize as airway hyperresponsiveness typical of asthma. Moreover, at end of the protocol, when they asked these subjects to take a deep breath, they proved to be refractory to the bronchodilator effect of a deep inspiration, just like asthmatics. The ability of these normal subjects to flip the states of their airways merely by including or precluding deep inspiration was consistent with the notion that these are conditionally stable states.
The airway muscle is akin to a spinning top. As long as you keep pumping in energy, it will keep spinning. For the airway, subjected to force fluctuations associated with breathing, every time a breath comes along, it keeps myosin molecules cycling and puts them in the perturbed and melted state. Both the airway and the spinning top flirt with disaster and can collapse to a static equilibrium if let to run down.
This hypothesis points to two important determinants of excessive airway narrowing in airways hyperresponsiveness. The first we have discussed extensively, namely, reduced forced fluctuations acting on airway muscle. The other is increased crossbridge cycling rate. It turns out that if the intrinsic rate at which myosin cycles is increased, it is harder to perturb the reaction. It is possible that an intrinsically faster myosin cycling rate in asthmatic airway muscle may contribute to airway hyperresponsiveness. In fact, research mostly from the laboratory of Dr. Newman Stephens has confirmed that in persons with asthma, the crossbridge cycling rate in airway muscle is increased.
Mechanisms of Airway Hyperresonsiveness
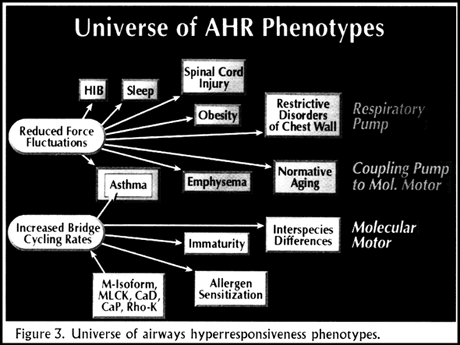
Airway hyperresponsiveness has been described in a wide variety of conditions. We will consider them in groups (Figure 3). Spinal cord injury, restrictive disorders of the chest wall, obesity, and sleep are conditions associated with an increased propensity for airways hyperresponsiveness or asthma. Each of these is a situation where one would expect reduced force fluctuations acting on airway muscle because of reduced chest wall excursion. In these circumstances airway hyperresponsiveness may be a manifestation of decreased activity of the respiratory pump.
The next group of conditions with an increased incidence of airway hyperresponsiveness includes emphysema, normative aging, and asthma. In these circumstances the problem may be an uncoupling of the respiratory pump to the molecular motor of the airway muscle. In emphysema lung parenchyma is destroyed with loss of airway tethering; in normative aging there is progressive loss of lung elastic recoil; and in asthma airway inflammation causes thickening of the peribronchial adventitia.
The final group includes asthma, immaturity of the lung, allergen sensitization, and inter-species differences. It turns out that in each of these situations, there is reason to believe that the intrinsic rate of crossbridge cycling is increased. Here the problem may be at the level of the molecular motor. It is running so fast, with such high crossbridge cycling rates, that we cannot perturb the myosin binding with normal force fluctuations. Based on this hypothesis, a number of other molecules suddenly become very interesting in the context of asthma. These include myosin isoforms, the amount and activity of myosin light chain kinase, accessory proteins such as caldesmon and calponin, known to regulate crossbridge cycling rates, and Rho-kinase.
Based on the perturbed equilibrium hypothesis that I have presented, one can take a diverse group of respiratory diseases that appear to be unrelated but that share a tendency toward airway hyperresponsiveness and suggest a unified framework for their understanding and further investigation.
About the author: Jeffrey J. Fredberg, Ph.D. is Professor of Bioengineering and Physiology and Director of the Physiology Program in the Department of Environmental Health at Harvard School of Public Health and Lecturer in Mechanical Engineering at the Massachusetts Institute of Technology. He has been a long-time collaborator in asthma research with members of the Pulmonary and Critical Care Division at Brigham and Women's Hospital, including Drs. Roland Ingram and Jeffrey Drazen.
References:
Fredberg JJ, Inouye DS, Mijailovich SM, Butler JP. Perturbed equilibrium of myosin binding in airway smooth muscle and its implications in bronchospasm. Am J Respir Crit Care Med 1999; 159:1-9.
Fredberg JJ. Frozen objects: small airways, big breaths, and asthma. J Allergy Clin Immunol 2000; 106:615-24.
Moore BJ, Verburgt LM, King GG, Pare PD. Effect of deep inspiration on methacholine dose-response curves in normal subjects. Am J Respir Crit Care Med 1997; 156:1278-81.
Asthma Grand Rounds Bulletin, published between 1997 and 2005, provided brief, written summaries of our early Asthma Grand Rounds presentations.